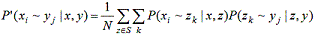Probabilistic Consistency-based Multiple Alignment of Amino Acid Sequences |
| RUN | ABOUT | ALGORITHM | DOWNLOAD | HELP | |||||||
|
PROBCONS is a consistency based multiple aligner. Traditional alignment methods based on edit distance score an alignment as the sum of similarity values for aligned residues and length dependent gap penalties for unaligned positions. Scoring a multiple alignment in a probabilistically rigorous and biologically motivated manner, and finding the optimal alignment once a scoring scheme has been specified, are not straightforward tasks. In practice, the ad hoc sum-of-pairs measure, which combines the projected pairwise distances of all pairs of sequences in the alignment are commonly used for scoring. Many heuristic strategies use progressive alignment based on an evolutionary tree or iterative approaches, but are prone to errors in early stages of alignment. To combat this, post-processing steps such as iterative refinement are applied. Consistency-based schemes take the alternative view that "prevention is the best medicine." For any multiple alignment, the induced pairwise alignments are necessarily consistent, i.e., given a multiple alignment of x, y, and z, if position xi aligns with position zk and position zk aligns with yj in the projected x-z and z-y alignments respectively, then, xi must align with yj in the projected x-y alignment. Consistency-based techniques apply this principle in reverse, using alignments to intermediate sequences as evidence to guide the pairwise alignment of x and y. This principle forms the basis of the PROBCONS aligner. Formally, the algorithm is described below:
Some of the steps are described in detail below: In the pairwise alignment case, an optimal expected accuracy alignment is calculated by applying the maximum weight trace algorithm to a matrix consisting of the P(xi ~ yj | x, y) values. For the multiple sequence case, we rely on a progressive alignment scheme using the guide tree calculated by hierarchical clustering of sequences. Initially, progressive alignment proceeds by assigning each sequence to its corresponding leaf in the tree. If a node has exactly two leaves as children, then the sequences assigned to the children are aligned, and this alignment is assigned to the node. Finally, the leaves of the node are removed so that the node becomes a leaf, and the process iterates until only a single node remains and all sequences have been aligned. When aligning groups of sequences, we use a sum-of-pairs scheme in which the score of a multiple alignment is given by summing the expected accuracy for each possible projection of the multiple alignment to two sequences. Thus, we can find the optimal expected accuracy alignment of a group of sequences by a straightforward extension of the pairwise dynamic programming computation. Guide trees are computed in a greedy hierarchical manner. Given a set S of sequences to be aligned, we denote the expected accuracy for aligning two sequences x and y as E(x, y). Initially, each sequence is placed in its own cluster. Then, the two clusters x and y with the highest expected accuracy are merged to form a new cluster xy; we then define the expected accuracy of aligning xy with any other cluster z as E(x, y)(E(x, z) + E(y, z)) / 2. This process is repeated until only a single cluster remains. The probabilistic consistency transformation When aligning two sequences x and y with a set of multiple sequences, S, we ideally would like to provide an estimate for P(xi ~ yj | S). In practice, we use the following heuristic decomposition: where we set P(xi ~ xj | x) to 1 if i = j and 0 otherwise. In this sense, the approximate probabilistic consistency calculation may be viewed as a transformation that, given a set of all-pairs pairwise posterior probabilities, produces a new set of all-pairs pairwise posterior probabilities that have been adjusted to account for a single intermediate sequence. By iterated applications of the transformation, then, we can approximate the effect of accounting for more than one intermediate sequence at a time. |